
二类医疗器械经营备案经营范围变更:审批难点科普
在现代商业环境中,企业运营常需应对各种法规调整,其中二类医疗器械经营备案的范围变更,便是许多企业主关注的焦点。
这类变更不仅涉及复杂的法规要求,还可能因细节疏忽导致审批延误,影响业务正常开展。
本文将深入探讨二类医疗器械经营备案范围变更的审批难点,帮助企业主更好地理解这一过程,从而高效应对挑战。
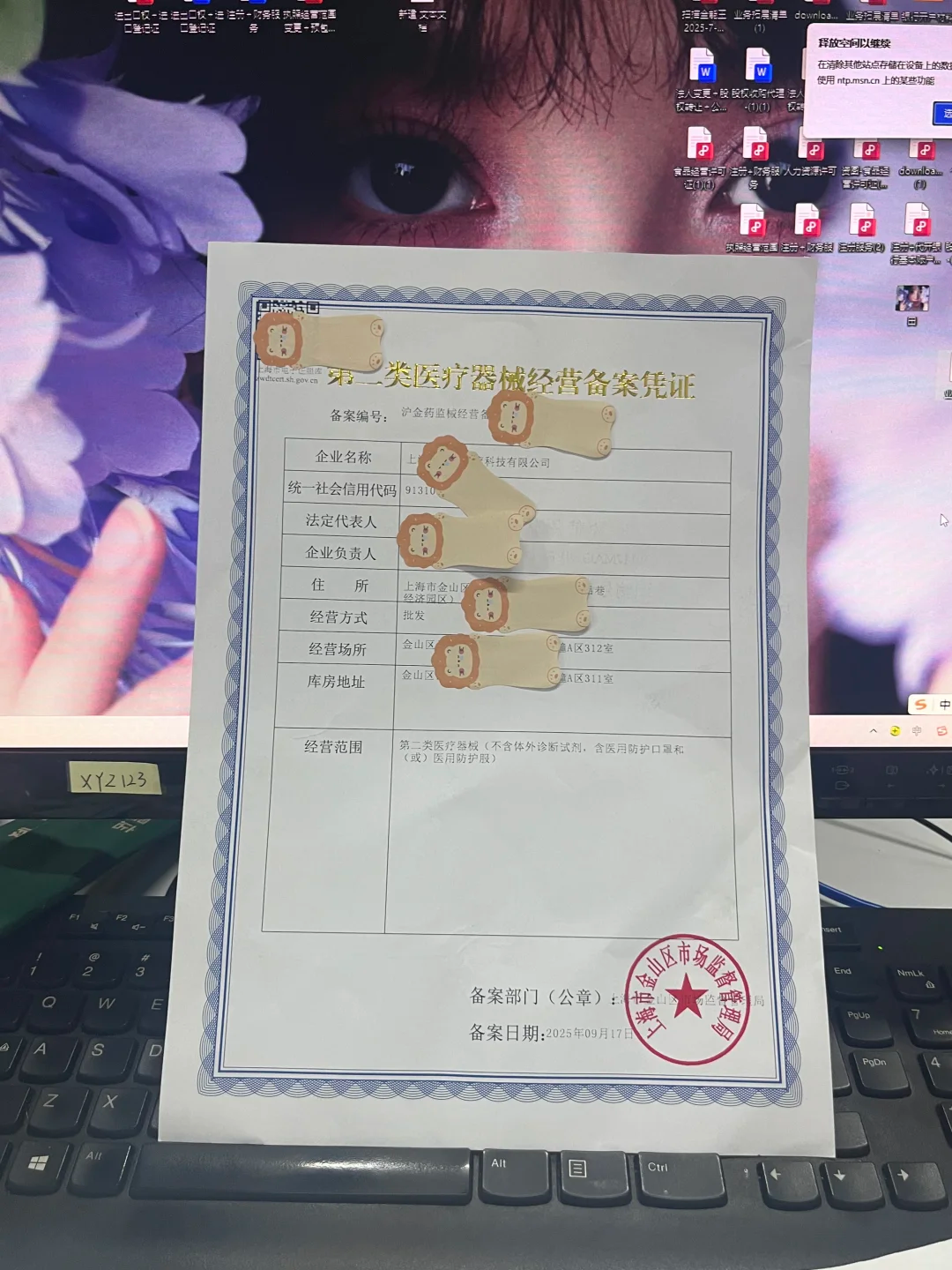
什么是二类医疗器械经营备案?
二类医疗器械通常指那些对人体有潜在风险、需严格控制管理的器械,如部分诊断设备或治疗器具。
经营备案是企业从事相关活动的法定前提,确保产品从生产到销售的每个环节都符合安全标准。
当企业业务扩展或调整时,例如新增产品线或变更服务模式,就需要对原有备案范围进行更新。
这一过程看似简单,实则涉及多方面的合规审查,稍有不慎便可能触发审批障碍。
审批过程中的主要难点
1. 法规理解偏差:二类医疗器械的监管框架较为细致,企业主往往因不熟悉较新政策而提交不完整的申请材料。
例如,备案范围需精确匹配产品分类,若描述模糊或超出原定类别,审批机构可能要求补充说明,导致时间拖延。
此外,法规更新频繁,企业若未及时跟进,容易沿用旧标准,造成申请被退回。
2. 材料准备不充分:备案变更需提供详实的证明文件,包括产品技术说明、质量管理体系记录等。
许多企业因内部管理松散,无法快速整理出符合要求的文档。
例如,若企业新增高风险器械,需额外提交安全评估报告,但若缺乏专业指导,往往忽略这一环节,引发审批停滞。
3. 流程衔接不畅:备案变更涉及多个环节的协同,从初审到复核,需确保信息一致。
企业常因部门沟通不足,导致提交的申请与实际情况不符。
例如,工商登记信息未及时更新,可能被认定为信息不实,进而影响整体进度。
这种内部协调问题,往往成为隐形的审批障碍。
4. 风险控制要求严格:二类医疗器械直接关系公共健康,审批机构会重点关注企业的风险管控能力。
如果企业未能展示完善的质量监督机制,例如缺乏定期培训记录或应急预案,审批可能被暂停。
这要求企业不仅注重材料形式,更需强化内部管理实践。
如何高效应对这些难点?
面对这些挑战,企业可采取系统性策略来优化备案变更过程。
首先,提前进行法规调研至关重要。
通过专业渠道获取较新政策解读,确保申请内容符合当前标准。
企业可建立内部合规小组,定期审核经营流程,防患于未然。
其次,注重材料准备的细节。

建议企业梳理现有文档体系,确保所有证明文件齐全、准确。
例如,对于新增产品,提前收集技术参数和测试报告,避免临时补漏。
同时,加强跨部门协作,确保工商、财务等信息同步更新,减少因信息不一致导致的返工。
此外,借助外部专业支持也能显著提升效率。
经验丰富的服务团队可提供针对性指导,帮助企业规避常见陷阱。
例如,他们能协助审核材料完整性,或模拟审批流程,提前发现潜在问题。
这不仅节省时间,还能降低因反复修改带来的成本。
最后,企业应强化内部风险管理。
通过定期员工培训和模拟审计,提升团队对法规的敏感度。
建立标准化操作流程,确保从采购到销售的每个环节都符合备案要求。
这种前瞻性管理,不仅能顺利通过审批,还能为企业长期发展奠定坚实基础。
结语
二类医疗器械经营备案的范围变更,虽充满挑战,但通过深入了解审批难点并采取有效措施,企业完全可以实现平稳过渡。

关键在于提前规划、注重细节,并善用专业资源。
作为深耕企业服务多年的团队,我们始终秉持诚信、专业和高效的理念,致力于为客户提供全方位支持,帮助您在合规道路上省心省力,专注核心业务发展。
如果您正面临类似问题,欢迎进一步交流,共同探索更优解决方案。
 319591724
319591724